
 |
| Rhinolophus affinis from Thailand. Photo by Charles Francis. |
Based on virus genome sequencing, bats have been mentioned in connection to the origin of SARS-CoV2 from the very beginning. This early picture then became more complicated as in depth-analyses revealed a key part of the viral genome that enables entry into host cells —the receptor binding domain (RBD) of the spike protein— is more closely related to similar domains from viruses found in pangolins. But the close evolutionary relatedness between the human pathogen (SARS-CoV2) and a virus not known to cause disease in humans (RatG2013, sampled in 2013 from the Intermediate horseshoe bat Rhinolophus affinis) is undisputed. Yet it’s difficult to parse out the wildlife source of SARS-CoV2 because despite its close relatedness (see below) to other wildlife viruses, it is not the direct result of recombination of any of the wildlife viruses known to date. A preprint by Boni et al. (2020) provides the evolutionary context for better understanding the wildlife origin of SARS-CoV.
Phylogenies, the evolutionary representation of relatedness among species can be hard to read. Below, is the phylogeny that Boni et al. (2020) estimated using the genomes of many different viruses sampled from wildlife. How closely related the different viruses are depends on how recently they share a common ancestor so that the red human (SARS-CoV2) and the bat virus (RatG2013) in the figure are sister lineages–as close as any two separate branches can get. But this phylogeny provides even more information: the length of the branches is proportional to time, so it can actually tell us when the bat virus and the human pathogen went their separate ways or diverged. This is why the inset shows several different sets of dates depending on what viruses are compared; however SARS-CoV2 and the bat virus RatG2013 are separated by 40-70 years! In other words, there is no way the human pathogen came directly from the virus that was circulating among intermediate horseshoe bats in 2013. Thanks to these genomic analyses we know that to figure out where SARS-CoV2 really came from in 2019, we have to keep looking at other possible hosts, and in the wild.
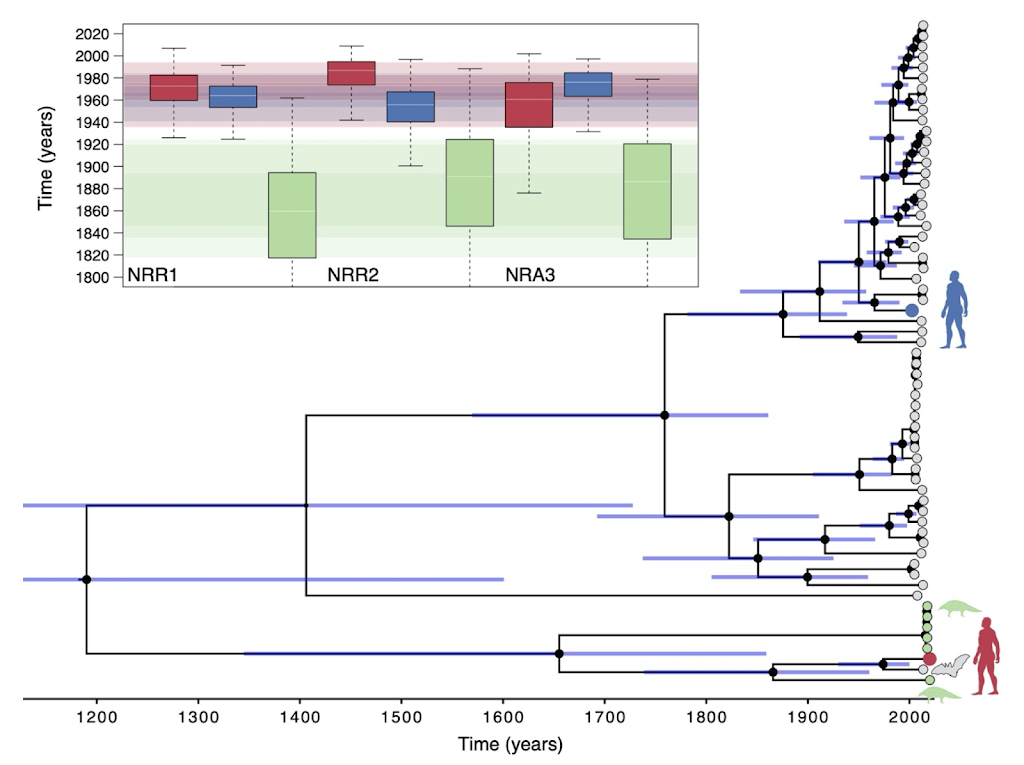 |
| Timetree of sarbecovirus lineages. The boxplots represent the divergence time estimates for SARS-CoV-2 (red boxplot) and the 2002-2003 SARS-CoV virus (blue boxplot) from their most closely related bat virus. Green boxplots show the time to most recent common ancestor estimate for the RaTG13/SARS-CoV-2 lineage and its most closely related pangolin lineage (from Guangdong 2020). Grey tips correspond to bat viruses, green to pangolin, blue to the SARS-CoV virus, and red to SARS-CoV-2. The size of the black internal node circles are proportional to the posterior node support. 95% credible intervals bars are shown for all internal node ages. Legend modified from Boni et al. 2020. |
Source: Boni, M. F., P. Lemey, X. Jiang, T. T.-Y. Lam, B. Perry, T. Castoe, A. Rambaut et al. 2020. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. bioRxiv:2020.2003.2030.015008.